Hydrogen (Proton, Deuterium and Tritium) NMR
Use our NMR service that provides 1H NMR and many other NMR techniques.
There are three isotopes of hydrogen used in NMR spectroscopy: 1Hydrogen, 2Deuterium and 3Tritium. Each isotope resonates at a very different frequency for example if 1H resonates at 400 MHz then 2H resonates at 61.402 MHz. Only one isotope is observed at a time because the spectrometer transmits and receives over a very limited frequency range. The chemical shift ranges for all three nuclei are virtually identical and can be used for preliminary analysis but there the similarity ends. 3Tritium is not commonly measured by NMR because it is radioactive.
Each type of signal has a characteristic chemical shift range (fig. 1) that can be used for initial assignment.
Fig. 1. Chemical shift ranges of protons according to their chemical environment
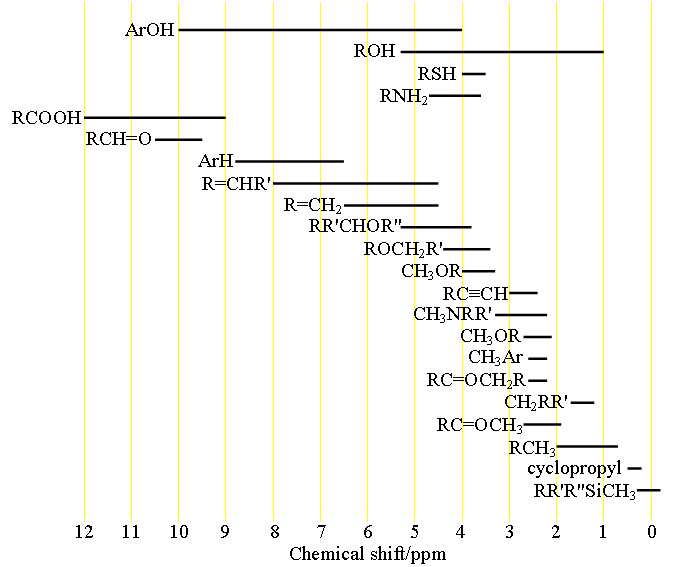
Choose the structure that most closely represents the hydrogen in question. R = alkyl or H, Ar = aryl.
1Hydrogen (Proton) NMR
The 1D 1H (Proton) NMR experiment is the most common NMR experiment. The proton (1Hydrogen nucleus) is the most sensitive nucleus (apart from tritium) and usually yields sharp signals. Even though its chemical shift range is narrow, its sharp signals make proton NMR very useful. Our NMR service provides proton NMR along with many other NMR techniques.
A typical analysis of a 1H NMR spectrum may proceed as follows:
The number of protons of each type in the spectrum of a pure sample can be obtained directly from the integrals of each multiplet. This is only true if the multiplets are well separated and do not overlap the solvent or residual water signals and provided that the molecule is not undergoing slow conformational exchange. A routine NMR spectrum yields integrals with an accuracy of +/-10%. Accuracies of +/-1% can be achieved by increasing the relaxation delay to five times the longitudinal relaxation time (T1) of the signals of interest. Where multiplets overlap, the total integral of the spectral region may be used.
From the table of the proton chemical shifts one obtains information about each type of proton and can carry out a preliminary assignment.
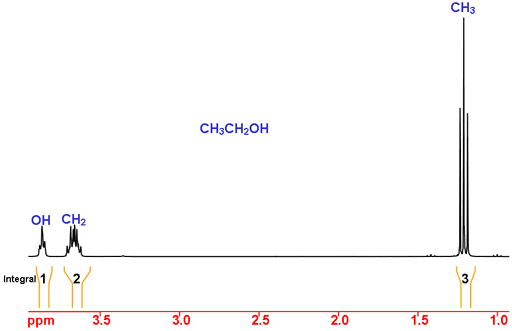
Consider ethanol as an example (Fig. 2). Using chemical shifts, the peak at 1.2 ppm is in the expected range for CH3 (methyl) and at 3.7 and 3.9 ppm are compatible with CH2 (methylene). The chemical shift of OH is very dependent on solvent and other experimental conditions so cannot be assigned by chemical shift alone.
Using integration, we expect CH3 to have an integral of three, CH2 to have an integral of two and OH to have an integral of one. This is the case and so the assignment is complete.
Fig. 2. 1H NMR spectrum of ethanol in CDCl3
For other molecules this is not sufficient and the multiplet structure is needed to complete the assignment. The multiplets (fig. 3) arise from spin-spin couplings that are transmitted through chemical bonds and yield information about the immediate molecular environment. In the case of CH3 and OH, they are split by the two neighboring protons of the CH2 to yield a triplet pattern called AX2. (Click here to see a list of common homonuclear splitting patterns and a description of heteronuclear coupling.) The CH2 is split by the single OH proton and the three CH3 protons to form the AX3Y pattern. (Click here for further examples of spectral assignment, 2D assignment of 12,14-ditbutylbenzo[g]chrysene and 2D assignment of cholesteryl acetate.)
Fig. 3. Multiplet structures from the 1H NMR spectrum of ethanol in CDCl3
Having determined the multiplicity, one may measure the coupling constants. These are measured in Hz (not ppm), as they are field independent. If you find that two (and only two) multiplets contain the same coupling constant then you know that they arise from nearby protons. The coupling constant gives an indication of the distance between the protons. In general 10 to 18 Hz means two-bond or three-bond trans to a C=C double-bond. Coupling constants between 1 to 10 Hz indicate three-bonds or more bonds if delocalized. Less than 1 Hz usually means four or more bonds.
In addition to homonuclear couplings, the multiplets may be split by other nuclei such as 19Fluorine or 31Phosphorus. (If such heteronuclear couplings are undesirable they may be decoupled. The best pulse sequence in such a case is that for inverse gated decoupling.)
Properties of 1H
| Property | Value |
|---|---|
| Spin | ½ |
| Natural abundance | 99.9845% |
| Chemical shift range | 13 ppm, from -1 to 12 |
| Frequency ratio (Ξ) | 100.000000% |
| Reference compound | TMS < 1% in CDCl3 = 0 ppm |
| Linewidth of reference | 0.08 Hz |
| T1 of reference | 14 s |
| Receptivity rel. to 1H at natural abundance | 1.000 |
| Receptivity rel. to 1H when enriched | 1.000 |
| Receptivity rel. to 13C at natural abundance | 5870 |
| Receptivity rel. to 13C when enriched | 5871 |
2Deuterium NMR
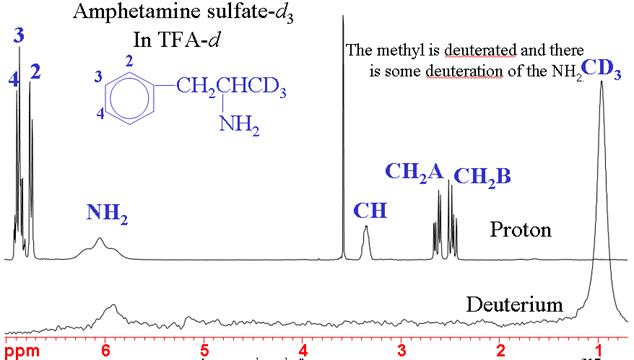
2Deuterium (heavy hydrogen) NMR is usually used for field frequency lock. At natural abundance it has very low sensitivity but when enriched it is of medium sensitivity. Deuterium usually yields broad signals whose line-width typically varies between a few hertz and a few kilohertz. The spectrum has the same narrow chemical shift range as for 1H but its low resolution and lower sensitivity make it a poor alternative. Deuterium-deuterium couplings are about 40 times smaller than proton-proton couplings and are therefore not observed. However, in partially deuterated molecules small proton-deuterium couplings can be observed. The main use of deuterium spectra is for determining the effectiveness of chemical deuteration (fig. 4).
Fig. 4. 1H and 2H NMR spectra of Amphetamine sulfate–d3 showing successful specific deuteration of the methyl
Properties of 2H
| Property | Value |
|---|---|
| Spin | 1 |
| Natural abundance | 0.0155% |
| Chemical shift range | 13 ppm, from -1 to 12 |
| Frequency ratio (Ξ) | 15.350609% |
| Reference compound | TMS-d12 neat = 0 ppm |
| Linewidth of reference | 1.7 Hz |
| T1 of reference | 1 s |
| Receptivity rel. to 1H at natural abundance | 1.50 × 10-6 |
| Receptivity rel. to 1H when enriched | 9.65 × 10-3 |
| Receptivity rel. to 13C at natural abundance | 8.78 × 10-3 |
| Receptivity rel. to 13C when enriched | 56.7 |
| Linewidth parameter | 0.41 fm4 |
3Tritium NMR
3T is the only nucleus more sensitive than proton (1H). Being a spin-½ isotope of hydrogen, spectra of fully tritiated compounds look similar to 1H with effectively the same chemical shifts but with slightly higher sensitivity, dispersion and coupling constants. However, 3T is very radioactive so most NMR studies are carried out with partially and specifically labeled compounds.
Our laboratory does not have the equipment to handle tritium or acquire tritium NMR spectra. However, we are willing to discuss the logistics if there is a demand for this service.
Properties of 3H
| Property | Value |
|---|---|
| Spin | ½ |
| Natural abundance | 0.0000000000000003% |
| Chemical shift range | 13 ppm, from -1 to 12 |
| Frequency ratio (Ξ) | 106.663974% |
| Reference compound | TMS-t1 < 1% in CDCl3 = 0 ppm |
| Linewidth of reference | ~0.1 Hz |
| T1 of reference | ~20 s |
| Receptivity rel. to 1H at natural abundance | 4 × 10-18 |
| Receptivity rel. to 1H when enriched | 1.21 |
| Receptivity rel. to 13C at natural abundance | 2 × 10-14 |
| Receptivity rel. to 13C when enriched | 7103 |
Safety note:
Some of the materials mentioned here are very dangerous. Ask a qualified chemist for advice before handling them. Qualified chemists should check the relevant safety literature before handling or giving advice about unfamiliar substances. NMR solvents are toxic and most are flammable. Specifically, TMS is toxic, volatile and flammable: wear protective gloves and work in a hood. All deuterated compounds are toxic. Tritium is radioactive: ensure that you have the necessary expertise and equipment before handling it.